Ribo-DB
Website
The current web browser interface allows to compose a complex query and to retrieve corresponding sequences..
IMPORTANT: the current DB is RiboDB/TEALS and is not the published initial DB I have not get tiome to modify this text, sorry.
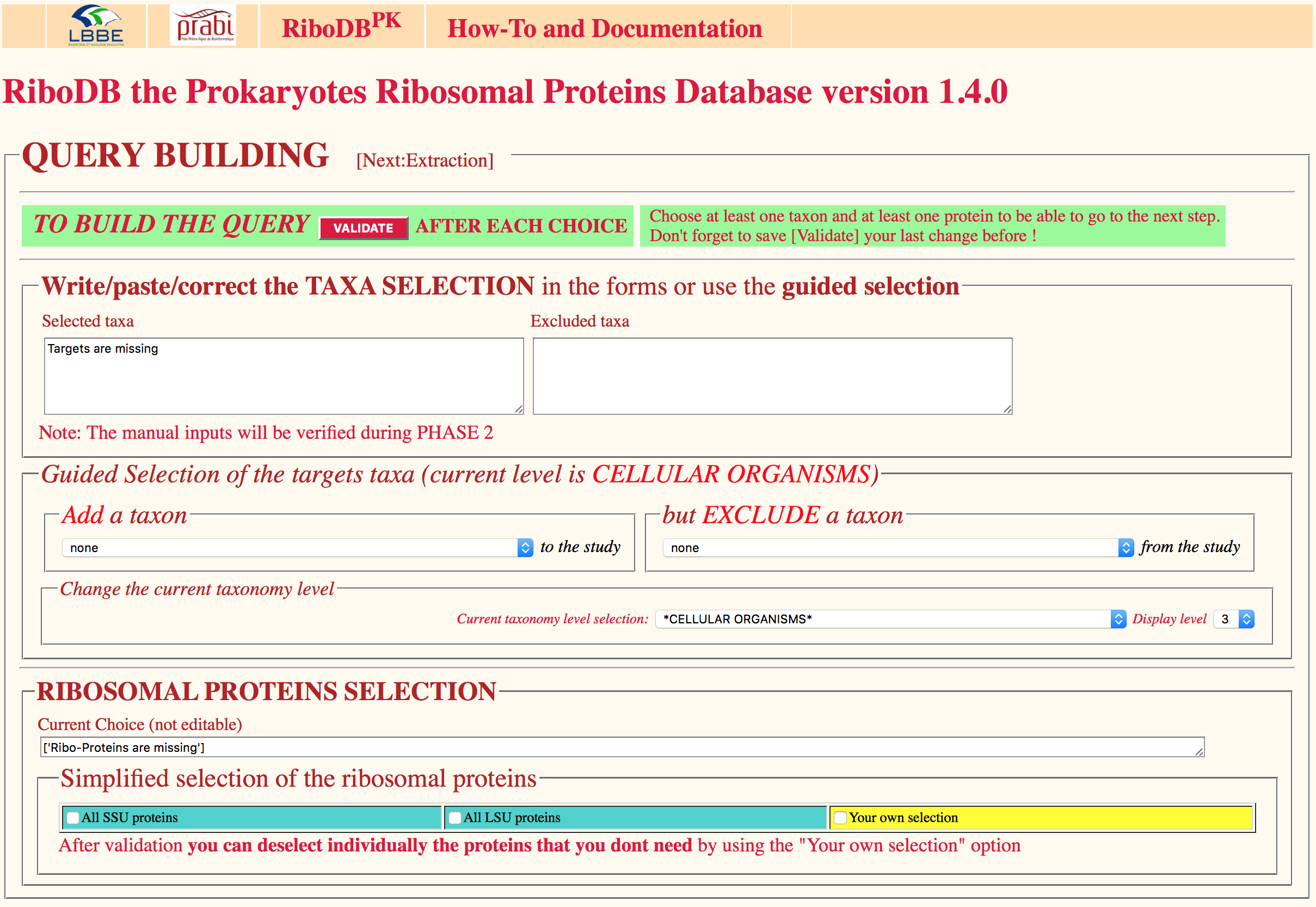
The dedicated RiboDB web browser interface allows users to extract nucleotide and/or amino acid r-protein sequences for a given set of strains. Selection of any taxonomic level among the Archaea and Bacteria is possible as well as any combination or exclusion of taxonomic levels. leBIBI-QBPP interrogation tool enables the submission of sequences and full automated phylogenetic positionning.
Navigation through the procaryotic taxonomy tree is possible prior the selection, but the desired taxonomy levels can be submitted also in the interface. This taxonomy browser relies on an ACNUC taxonomy server linked to the riboDB database.
The selection of any of the 90 ribosomal proteins, a given subset (small or large subunit of the ribosome) or even all of them is directly possible. A result page shows the evolution of the queries and of the extraction process.
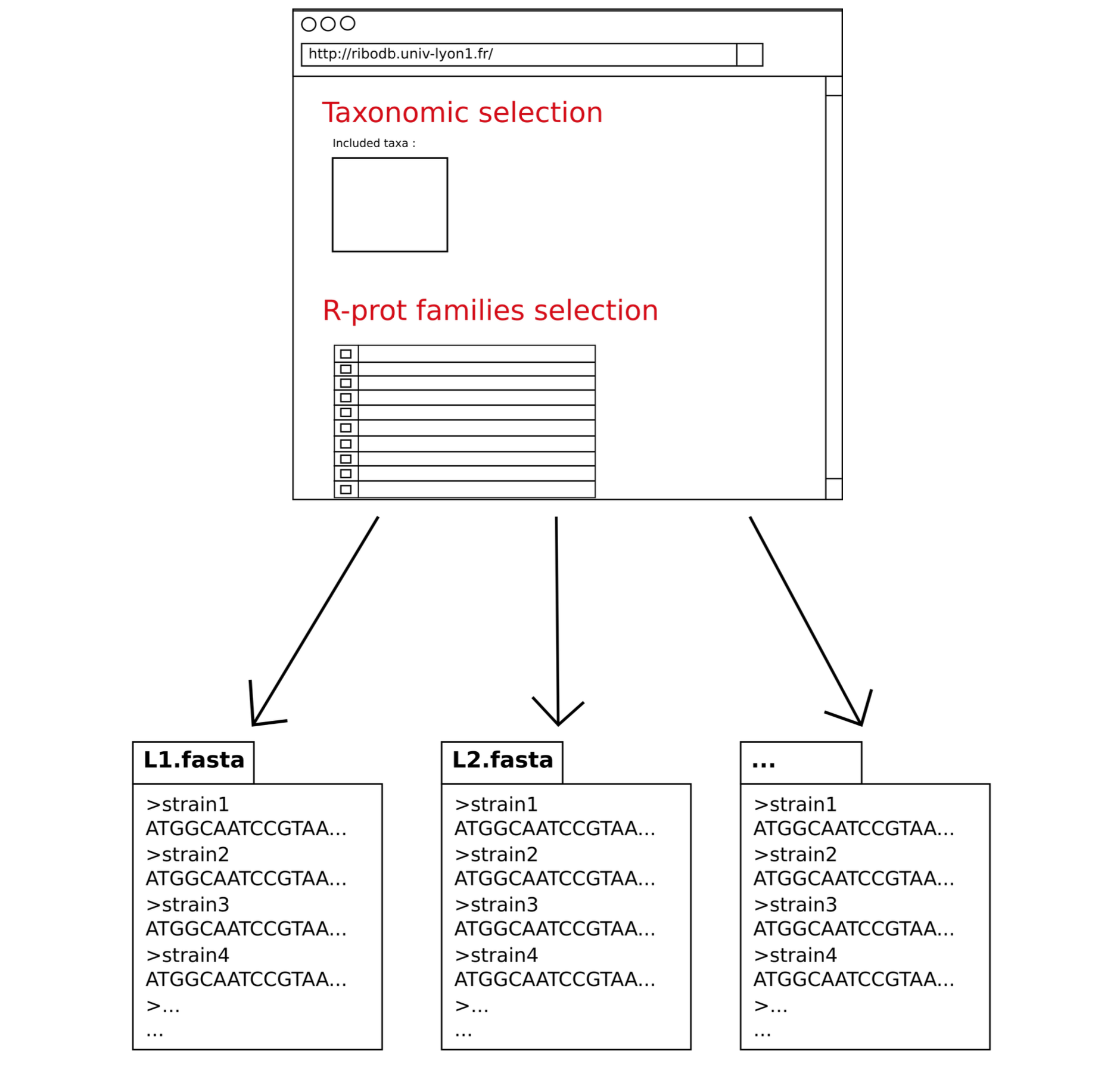
The final result is a set of fasta files (nucleotide and protein sequences) corresponding to the selected ribosomal proteins organized in directories. Directories or files can be browsed and dowloaded.
 The datasets curated for paralogs for use in phylogeny programs are also available. A compacted archive is also available.
The FASTA commentary line condenses all the main descriptors of a ribosomal protein sequence (species, strain, assemblyID, bankID, position, strand, taxonomic hierarchy) in a structured way.
The datasets curated for paralogs for use in phylogeny programs are also available. A compacted archive is also available.
The FASTA commentary line condenses all the main descriptors of a ribosomal protein sequence (species, strain, assemblyID, bankID, position, strand, taxonomic hierarchy) in a structured way.
The website is organized as a classical client-server system that uses python CGI to communicate. The python classACNUC is managing the interface to the ACNUC query language and retrieval capability.
BIBI-DB webtool
The analysis process
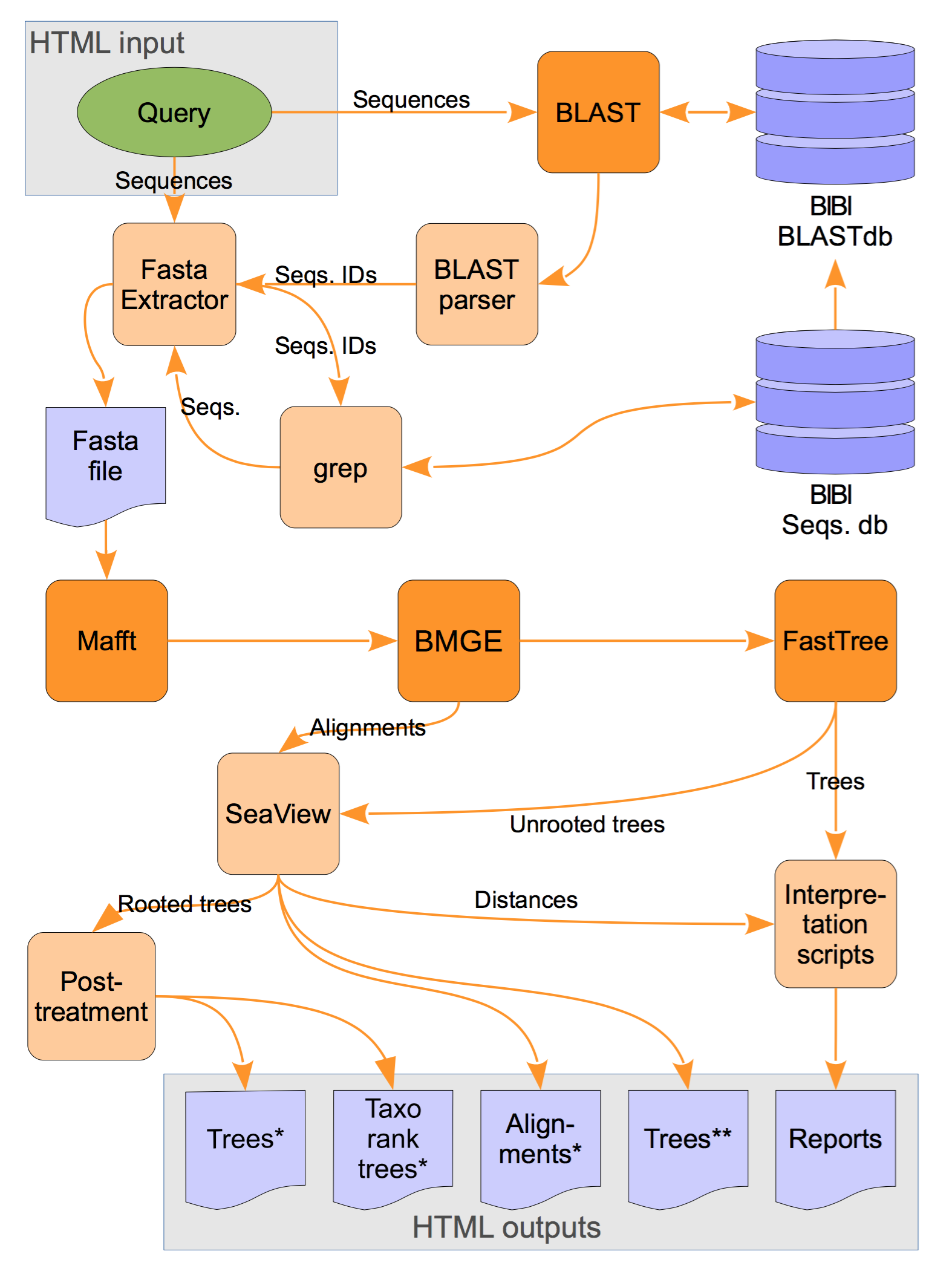
Global organization of the LeBIBIQBPP pipeline. BLASTN is run with the query against the selected database. A set of sequences with the highest similarity scores is extracted from the database. Selected sequences are multiply aligned by MAFFT the BMGE program is used to trim sequences and FastTree is then used to reconstruct the tree by maximum likelihood. Python scripts are analysing the results and organizing the graphical outputs generated by SeaView (see the bottom of this page for the softwares links). Sequence retrieval
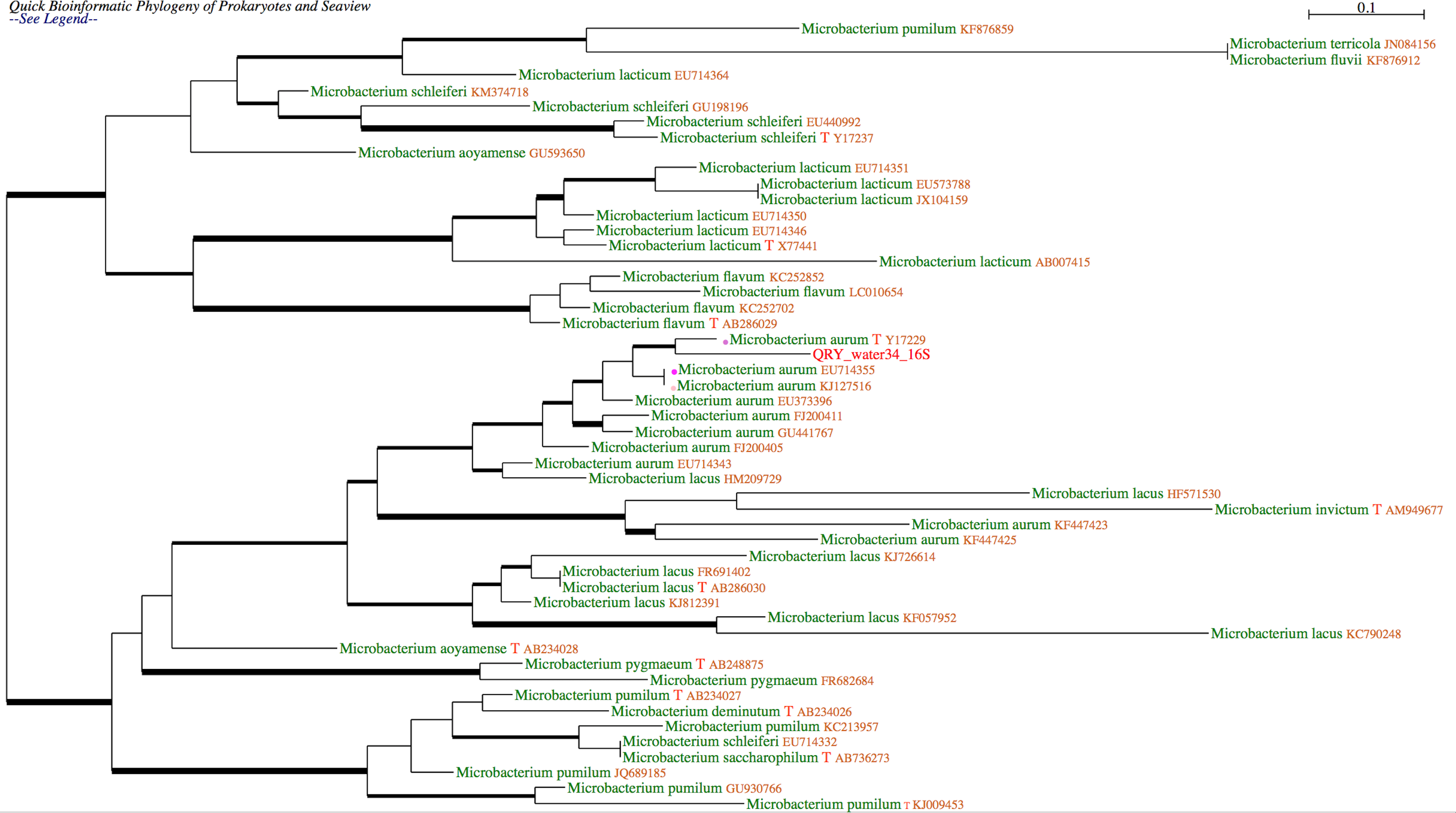
Example of tree
Phylogenetic placement of an undescribed bacterial sequence using another gene and database. The query sequence is suspected to be a new species of Mycobacterium. This was confirmed by the analysis of the rpoB sequence obtained from the same bacterial extract and the rpoB “stringent” database